Exploiting Pretrained Biochemical Language Models for Targeted Drug Design
This article has been accepted for publication in Bioinformatics Published by Oxford University Press. Preprint will be updated after publication.
Abstract
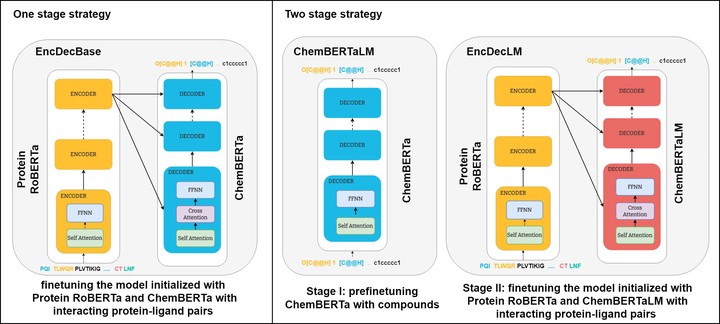
Motivation: The development of novel compounds targeting proteins of interest is one of the most important tasks in the pharmaceutical industry. Deep generative models have been applied to targeted molecular design and have shown promising results. Recently, target-specific molecule generation has been viewed as a translation between the protein language and the chemical language. However, such a model is limited by the availability of interacting protein–ligand pairs. On the other hand, large amounts of unlabelled protein sequences and chemical compounds are available and have been used to train language models that learn useful representations. In this study, we propose exploiting pretrained biochemical language models to initialize (i.e. warm start) targeted molecule generation models. We investigate two warm start strategies: (i) a one-stage strategy where the initialized model is trained on targeted molecule generation and (ii) a two-stage strategy containing a pre-finetuning on molecular generation followed by target-specific training. We also compare two decoding strategies to generate compounds: beam search and sampling. Results: The results show that the warm-started models perform better than a baseline model trained from scratch. The two proposed warm-start strategies achieve similar results to each other with respect to widely used metrics from benchmarks. However, docking evaluation of the generated compounds for a number of novel proteins suggests that the one-stage strategy generalizes better than the two-stage strategy. Additionally, we observe that beam search outperforms sampling in both docking evaluation and benchmark metrics for assessing compound quality.
Availability and implementation: The source code is available at https://github.com/boun-tabi/biochemical-lms-for-drug-design and the materials (i.e., data, models, and outputs) are archived in Zenodo at https://doi.org/10.5281/zenodo.6832145. We also provide a demo application where you can generate molecules right away at https://huggingface.co/spaces/gokceuludogan/WarmMolGen.
Gökçe Uludoğan
Teaching Assistant and PhD Student at Computer Engineering
My research interests include deep learning, cheminformatics and natural language processing.